Read Filters¶
Which reads are excluded before allele classification — the filter cascade, CLI flags, and defaults.
Visual Overview
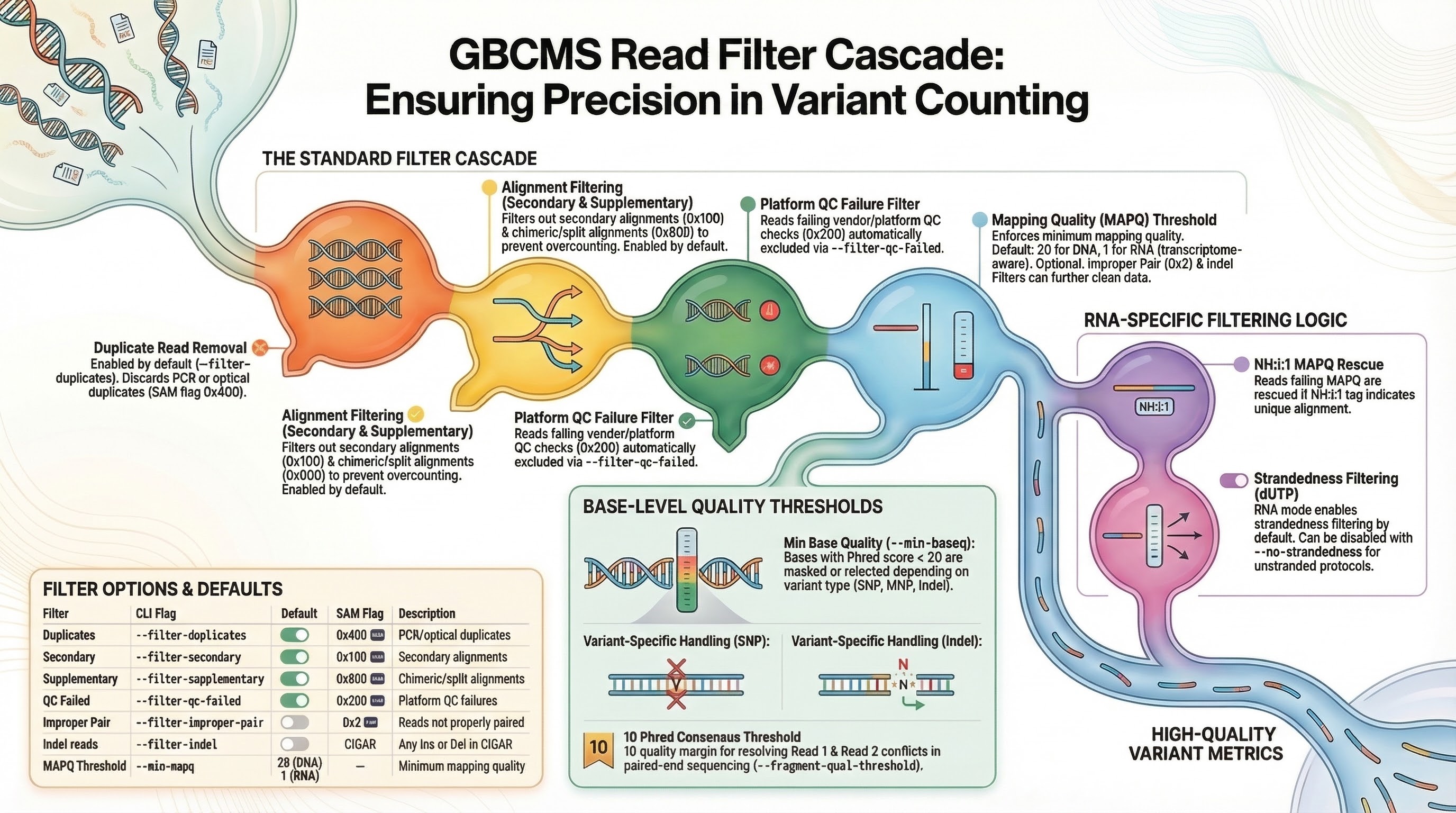
Filter Cascade¶
Every read from the BAM passes through a filter cascade before being checked for allele support. Reads failing any enabled filter are discarded. The order matches the Rust engine implementation.
flowchart LR
Start(["📖 BAM Read"]):::start
subgraph Auto ["🟢 Auto-On (default enabled)"]
direction LR
F1{"① Duplicate?\n0x400"}:::on
F2{"② Secondary?\n0x100"}:::on
F3{"③ Supplementary?\n0x800"}:::on
F4{"④ QC Failed?\n0x200"}:::on
F5{"⑤ MAPQ < threshold\n(20 DNA / 1 RNA)"}:::on
F1 -->|No| F2
F2 -->|No| F3
F3 -->|No| F4
F4 -->|No| F5
end
subgraph Opt ["⚙️ Optional (off by default)"]
direction LR
O1{"⑥ Improper pair?\n--filter-improper-pair"}:::off
O2{"⑦ Contains indel?\n--filter-indel"}:::off
O1 -->|No| O2
end
Start --> F1
F5 -->|Pass| O1
O2 -->|Pass| Done(["✅ Allele Classifier"]):::pass
F1 -->|Yes| Drop(["❌ Discard"]):::drop
F2 -->|Yes| Drop
F3 -->|Yes| Drop
F4 -->|Yes| Drop
F5 -->|Below threshold| Drop
O1 -->|"Yes (if enabled)"| Drop
O2 -->|"Yes (if enabled)"| Drop
classDef start fill:#9b59b6,color:#fff,stroke:#7d3c98,stroke-width:2px;
classDef on fill:#27ae60,color:#fff,stroke:#1e8449,stroke-width:2px;
classDef off fill:#95a5a6,color:#fff,stroke:#7f8c8d,stroke-width:2px;
classDef pass fill:#27ae60,color:#fff,stroke:#1e8449,stroke-width:2px;
classDef drop fill:#e74c3c,color:#fff,stroke:#c0392b,stroke-width:2px;
Filter Options¶
| Filter | CLI Flag | Default | SAM Flag | Description |
|---|---|---|---|---|
| Duplicates | --filter-duplicates |
On | 0x400 |
PCR/optical duplicates |
| Secondary | --filter-secondary |
On | 0x100 |
Secondary alignments |
| Supplementary | --filter-supplementary |
On | 0x800 |
Chimeric/split alignments |
| QC Failed | --filter-qc-failed |
On | 0x200 |
Platform QC failures |
| Improper Pair | --filter-improper-pair |
Off | 0x2 (inverted) |
Reads not properly paired |
| Indel reads | --filter-indel |
Off | CIGAR-based | Any Ins or Del in CIGAR |
| MAPQ threshold | --min-mapq |
20 (DNA) / 1 (RNA) | — | Minimum mapping quality |
Quality Thresholds¶
Beyond the read-level filters above, gbcms also applies base-level quality thresholds during allele classification:
| Parameter | CLI Flag | Default | Description |
|---|---|---|---|
| Min base quality | --min-baseq |
20 | Bases below this are masked or rejected |
| Fragment consensus threshold | --fragment-qual-threshold |
10 | Quality margin for R1/R2 conflict resolution |
How Base Quality Is Used
The effect of --min-baseq varies by variant type:
- SNP: Read is rejected if the base at the variant position has quality < threshold
- MNP: Read is rejected if any base in the MNP region has quality < threshold
- Insertion/Deletion (windowed scan): Inserted/deleted bases below threshold are masked (treated as wildcards) rather than rejecting the entire read
- Complex (Phase 2): Low-quality bases are masked — they cannot vote for either allele
- Complex (Phase 3 SW): Low-quality bases are replaced with
N, which scores 0 against any base
Comparison with Original GBCMS¶
Filter Non-Primary
The original GBCMS has a single --filter_non_primary flag. gbcms splits this into --filter-secondary and --filter-supplementary for finer control. Both default to on in gbcms (stricter than the original behavior, which was off).
| Feature | Original GBCMS | gbcms |
|---|---|---|
| Base quality filtering | No threshold | Default --min-baseq 20 |
| Duplicate filtering | Optional | On by default |
| Non-primary filter | Single flag (default off) | Split: secondary + supplementary (both On) |
| QC-failed filter | Optional | On by default |
| Indel read filter | Optional | Optional (off by default) |
Fetch Window¶
gbcms fetches reads from a dynamic window around each variant position — not just at the exact position. The window defaults to ±5bp but scales with the variant's repeat span to capture indels that aligners shift beyond 5bp in long homopolymers and microsatellite regions. The formula is max(5, repeat_span + 2).
Variant: chr1:100 A→ATG (insertion, repeat_span=0)
BAM fetch window: [95, 106) ← ±5bp (default)
Variant: chr1:200 A→AT (insertion in 15bp poly-A, repeat_span=15)
BAM fetch window: [183, 206) ← ±17bp (repeat_span + 2)
Anchor Overlap Gate
Although reads are fetched from the wider window, only reads that overlap the variant's anchor position (or are classified as REF/ALT via shifted indel matching) contribute to the DP count. This prevents DP inflation from reads that were brought in by the window padding but don't actually cover the variant.
RNA-Specific Filters¶
RNA mode (gbcms rna) extends the standard filter cascade with two additional checks.
NH:i:1 MAPQ Rescue¶
STAR assigns MAPQ=255 to uniquely mapped reads and MAPQ=0–3 to multi-mappers. When --min-mapq 1 (RNA default), reads that fail the MAPQ threshold are checked for the NH:i:1 tag (Number of Hits = 1). If present, the read is uniquely aligned and rescued.
flowchart TD
Read(["📖 RNA Read"])
Read -->|"read.mapq"| MAPQ{"MAPQ ≥ min_mapq?\n(default: 1)"}
MAPQ -->|"Yes — MAPQ=255\nunique alignment"| Pass(["✅ Pass"]):::pass
MAPQ -->|"No — MAPQ=0\n(check NH tag)"| NH{"NH:i:1?"}
NH -->|"Yes — novel junction\nunique but low MAPQ"| Rescue(["✅ Rescued"]):::rescue
NH -->|"No — NH>1\nmulti-mapper"| Drop(["❌ Discard"]):::drop
classDef pass fill:#27ae60,color:#fff,stroke:#1e8449,stroke-width:2px;
classDef rescue fill:#f39c12,color:#fff,stroke:#d68910,stroke-width:2px;
classDef drop fill:#e74c3c,color:#fff,stroke:#c0392b,stroke-width:2px;
Biological Context
Novel splice junctions often receive low MAPQ because STAR hasn't observed the junction in its first-pass database. The NH:i:1 rescue ensures these uniquely mapped reads contribute to allele counts rather than being silently discarded.
Strandedness Filter¶
For dUTP-stranded RNA-seq libraries, reads are classified by their orientation relative to the gene strand annotation. Both sense and antisense reads are counted (they contribute to rna_sense_depth and rna_antisense_depth respectively), but only sense-strand reads contribute to the primary DP/RD/AD counts when --enforce-strandedness is enabled.
| Read Orientation | Gene Strand | Classification |
|---|---|---|
| Forward (R1) | + | Sense |
| Reverse (R1) | + | Antisense |
| Forward (R1) | − | Antisense |
| Reverse (R1) | − | Sense |
Tip
Disable strandedness filtering with --no-strandedness for unstranded RNA-seq libraries.
BAQ Quality Downgrade¶
When --apply-baq is enabled, base qualities near CIGAR indels and splice junctions (CIGAR N/RefSkip) are heuristically downgraded to prevent inflated quality scores from causing false-positive allele classifications.
Parameters (Internal Constants)¶
These are not configurable via CLI — they are fixed values derived from Li (2011):
| Constant | Value | Description |
|---|---|---|
BAQ_RADIUS |
5 bp | Bases within 5 bp of an indel or splice junction boundary are penalized |
BAQ_PENALTY |
20 | Subtracted from BQ (clamped to 0, never negative) |
The effective aggressiveness of BAQ is controlled by --min-baseq (default 20): a base with BQ 30 near a splice junction becomes BQ 10 after BAQ, which is below the default --min-baseq threshold and is therefore excluded from allele evidence.
Mode Defaults¶
| Mode | Default | CLI Override | Rationale |
|---|---|---|---|
gbcms dna |
off | --apply-baq to enable |
DNA BAMs typically go through BQSR or consensus calling, which already recalibrates BQ. Enabling BAQ on top may double-penalize. |
gbcms rna |
on | --no-baq to disable |
RNA BAMs typically do not go through BQSR or consensus calling. BAQ penalizes bases near splice junctions, mimicking GATK SplitNCigarReads' overhang clipping. |
When to Enable BAQ for DNA¶
Enable --apply-baq on gbcms dna when the upstream pipeline does not include:
- GATK BQSR (Base Quality Score Recalibration)
- Consensus-based deduplication (fgbio, Marianas, Gencore)
- Any other base quality recalibration step
In these "raw BQ" scenarios, bases near indels may retain inflated sequencer-assigned quality scores that lead to false-positive allele calls. BAQ applies a -20 penalty within 5 bp of indel boundaries to compensate.
Do Not Enable for Pre-Calibrated BAMs
If your DNA pipeline includes BQSR or consensus calling, leave BAQ off (the default). Applying BAQ on already-recalibrated BAMs may over-penalize, causing undercounting of legitimate variant evidence.
RNA: Splice Junction Penalty¶
In RNA mode, BAQ additionally penalizes bases near CIGAR N (RefSkip) operations — splice junctions. This is a lightweight alternative to running GATK SplitNCigarReads upstream. See RNA Splice-Junction Handling for details.
Related¶
- Allele Classification — How reads passing filters are classified
- Counting & Metrics — Read-level and fragment-level counts
- DNA CLI Reference — DNA parameter options
- RNA CLI Reference — RNA-specific parameter options
abbreviations